Supervised Peak Calling for ChIP-Seq
Many peak detection algorithms have been proposed for ChIP-seq data analysis, but it is not obvious which algorithm and what parameters are optimal for any given data set.
In this work, conducted by Toby Hocking, we propose a supervised approach that uses labels produced from visual inspection of the aligned read counts. The main idea is to manually label a small subset of the genome, and then learn a model that makes consistent peak predictions on the rest of the genome.
The main advantages of this approach are:
- An automatic optimization of the peak calling parameters.
- A seamless integration of the data to analyze (e.g. quality, signal/noise, narrow/broad profiles).
- A consistent and more interpretable calling across multiple samples
In practice, the user is presented with the read coverage profile in a few regions/samples and asked to add “labels”. These labels are then used to train the model and call peaks in the rest of the genome/samples.
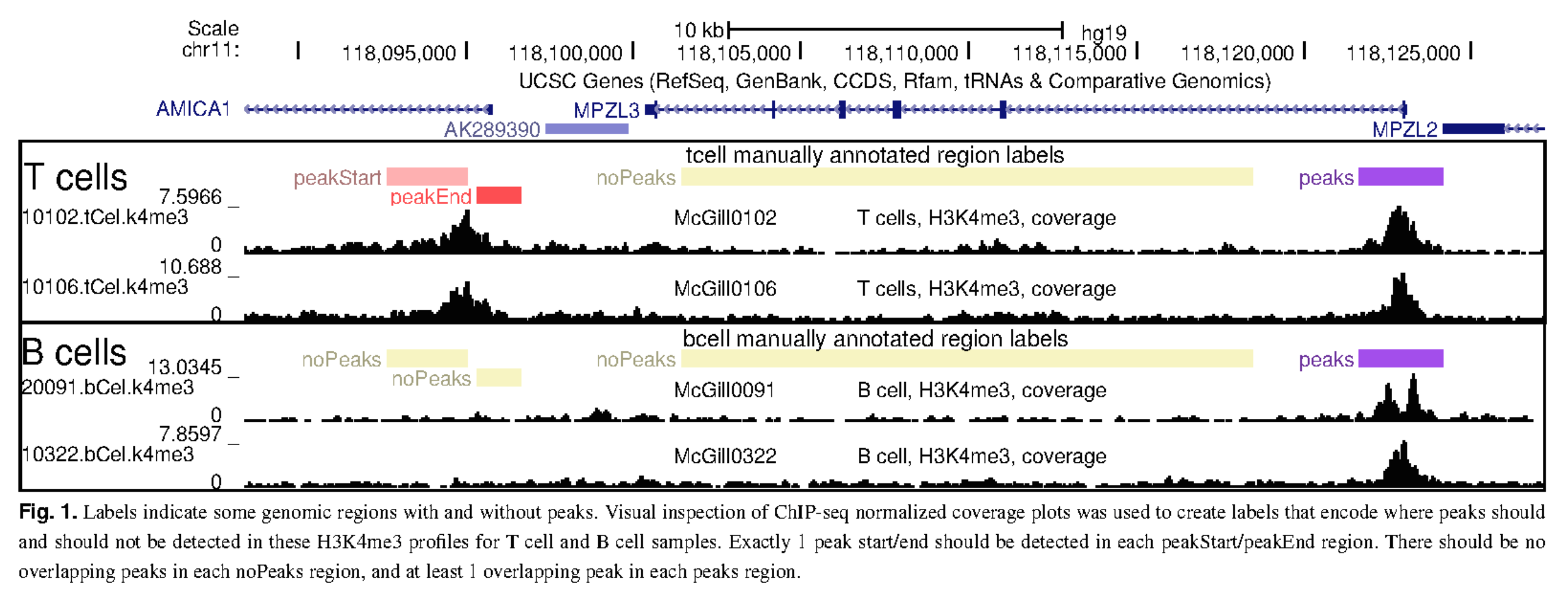
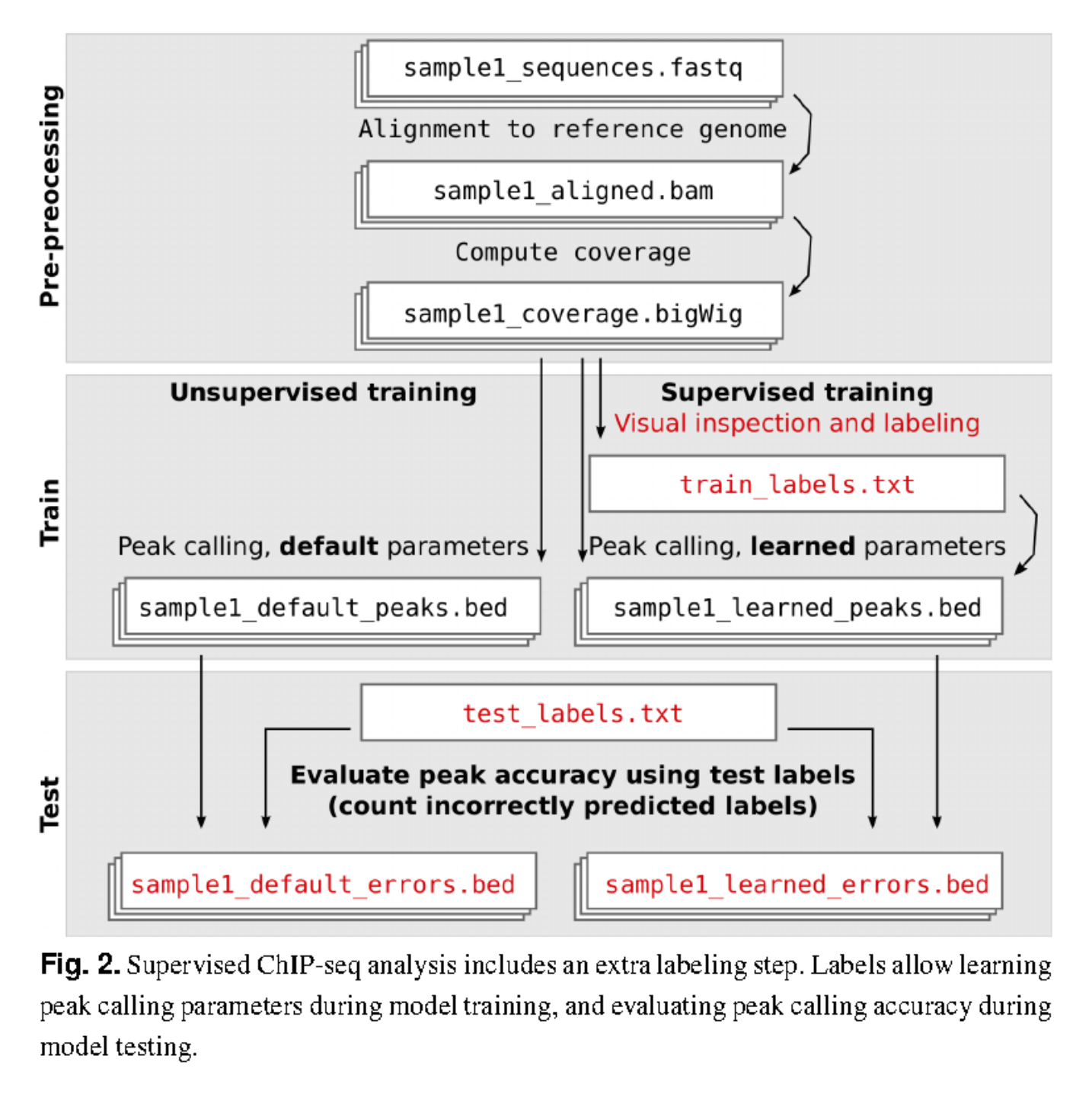
A labeling step is added to the analysis workflow.
Thanks to this visual inspection and labeling, the user can evaluate the peak calling accuracy of the calls, from both unsupervised and supervised calling.
Software & Publications
PeakError
Evaluate ChIP-Seq peaks accuracy using a set of labels.
PeakSegJoint
PeakSeq extension to jointly analyze several ChIP-Seq samples.