
Par David Lougheed
À propos des séquences répétées en tandem courtes
Début 2021, Jeffrey Hyacinthe, un autre étudiant du laboratoire de Guillaume Bourque, a écrit sur les séquences répétitives, en particulier sur les éléments transposables (ET). Je vais ici aborder un autre type de séquence répétitive : les séquences répétées en tandem courtes (STR), également appelées microsatellites. Contrairement aux ET, elles ne se déplacent pas et contiennent de multiples copies de courts motifs de 2 à 6 paires de bases (Gymrek et al., 2012 ; 2016). Le nombre de répétitions à un emplacement donné peut varier au sein d’un même individu ou d’une population. On estime à environ 700 000 le nombre de STR dans le génome humain (Gymrek et al., 2016), ce qui signifie qu’elles sont très répandues. Ils constituent le sujet principal de ma thèse de maîtrise, commencée l’automne dernier au laboratoire Bourque (http://www.computationalgenomics.ca/BourqueLab/).
Les STR ont été associés à divers phénotypes pathologiques, tels que la maladie de Huntington (MH) et le syndrome de l’X fragile (SXF), ainsi qu’à d’autres effets comme la variation d’expression (Gymrek et al., 2016). Dans la MH, une expansion de la répétition C-A-G dans le cadre de lecture du gène HTT codant pour la protéine déclenche l’apparition de la maladie avant l’âge de 30 ou 40 ans (MedlinePlus, 2020 ; voir tableau 1). La MH est associée à des troubles moteurs et cognitifs, et conduit finalement au décès. Une seule copie de l’allèle pathogène suffit à provoquer l’apparition de la maladie.
10-35Aucun phénotype40+Apparition quasi certaine de la maladie de Huntington
| Nombre de répétitions | Phénotype |
|---|---|
| 36-39 | Apparition potentielle de la maladie de Huntington |
Tableau 1 : Nombre de répétitions CAG dans le gène HTT (coordonnées GRCh38 : chr4:3074876-3074933) et les phénotypes associés. Voir MedlinePlus, 2020.
Les chercheurs ont consacré d’importants efforts à l’annotation des régions répétitives dans les génomes de référence actuels. L’outil Tandem Repeats Finder, initialement publié en 1999 par Gary Benson, est utilisé pour cataloguer les répétitions « simples » dans les génomes de référence. Ces annotations peuvent servir de cibles à d’autres outils pour le génotypage ou la détection d’expansions (Mousavi et al., 2019).
Malgré ces annotations, l’analyse de l’ADN répétitif reste complexe avec les technologies génomiques actuelles. En 2021, la plateforme de séquençage Illumina à courtes lectures (Amarasinghe et al., 2020) s’est imposée comme la norme. Elle produit généralement des lectures appariées d’une longueur maximale d’environ 150 paires de bases (Illumina, 2020). L’identification des régions répétitives plus longues pose un problème majeur (Figure 1).
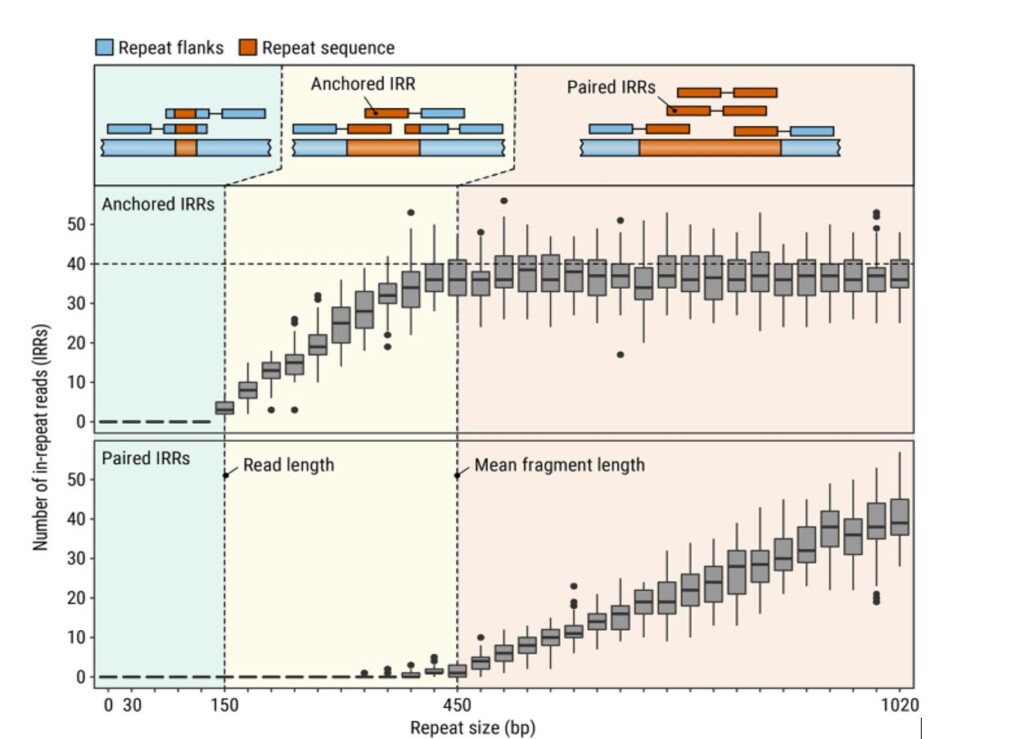
Figure 1 : Types de paires de lectures issues du séquençage Illumina et leur alignement sur des séquences répétitives de différentes tailles. D’après Dolzhenko et al., 2020. Ce schéma montre que pour les séquences répétitives de plus de 150 pb, les lectures flanquant les extrémités des deux séquences disparaissent, et il est alors nécessaire d’utiliser des méthodes approximatives basées sur les lectures intra-répétitives (IRR) pour déterminer le nombre de répétitions. Les IRR appariées peuvent potentiellement s’aligner sur plusieurs régions du génome de référence, puisqu’elles ne possèdent pas de séquence d’ancrage.
De longues séquences présentant le même motif n’apparaissent pas nécessairement de manière unique à une seule position du génome humain (Dolzhenko et al., 2017). Sans contexte plus large des régions environnantes, les séquences d’un locus peuvent potentiellement correspondre à d’autres loci, ce qui en réduit l’utilité.
De plus, étant principalement des organismes diploïdes – la plupart des loci répétitifs étant présents en deux copies dans le génome humain –, le processus d’identification est encore complexifié. Si ces loci sont étendus ou si la couverture de séquençage est insuffisante dans une zone donnée, l’identification peut devenir ambiguë et entraîner des pertes alléliques ou d’autres difficultés à déterminer un génotype précis (Gymrek et al., 2012 ; 2016 ; Mousavi et al., 2019).
D’autres techniques prometteuses pour le génotypage STR reposent sur des technologies de séquençage à longues lectures telles que le séquençage Oxford Nanopore (ONT) ou le séquençage Pacific Biosciences (PacBio) SMRT. Les séquences longues, généralement de l’ordre du kilobase (Amarasinghe et al., 2020), fournissent des informations sur la structure globale de chaque séquence et éliminent les problèmes d’alignement mentionnés précédemment, sauf dans des cas extrêmes. Cependant, cela se traduit par des coûts de séquençage plus élevés, une couverture généralement plus faible et des taux d’erreur plus importants (Amarasinghe et al., 2020). Relativement peu d’outils ont été développés pour le génotypage ciblé et le criblage pangénomique des STR à partir de données de séquençage long.
Technologies de détection des STR
Des outils spécifiques permettent de profiler la variation des STR et de déterminer les génotypes. Pour déterminer un génotype STR, on utilise généralement le nombre de répétitions du motif pour représenter un allèle particulier. Par exemple, pour la répétition HTT, un génotype 20/35 correspond à 20 et 35 copies de CAG, soit 60 et 105 bases, sur les deux copies du chromosome 4 chez l’individu.
Certaines des premières méthodes bioinformatiques fonctionnant sur des données NGS ont été publiées au début des années 2010. L’un des premiers exemples est lobSTR (Gymrek et al., 2012), conçu pour les données de séquençage à haut débit de l’époque et validé par électrophorèse sur gel d’ADN. Parmi les autres outils dédiés aux STR apparus au cours de la dernière décennie, on peut citer hipSTR, Tredparse et STRetch. Ces méthodes s’appuient sur des catalogues et ne peuvent généralement pas détecter le nombre de répétitions des STR plus longs qu’une lecture Illumina, ou ne signalent que les expansions (Mousavi et al., 2019).
Plus récemment, des outils comme ExpansionHunter (EH ; Dolzhenko et al., 2017) et gangSTR (Mousavi et al., 2019) ont développé de nouvelles techniques permettant d’obtenir un plus grand nombre d’appels de meilleure qualité, des performances accrues et la possibilité de calculer des intervalles de confiance pour des régions répétées plus longues que les lectures elles-mêmes. Ces techniques utilisent une combinaison de lectures ancrées/flancantes et de répétitions intra-lectures (voir Figure 1) pour identifier précisément les STR courts et les régions STR longues de manière plus approximative.
Une approche alternative utilisant des lectures courtes pour analyser les régions STR longues a été introduite par les auteurs d’EH en 2020 sous le nom d’ExpansionHunter Denovo (EHDn ; Dolzhenko et al., 2020). Cet outil n’est pas un détecteur de séquences répétées, car il ne cherche pas à déterminer le nombre de répétitions à un locus donné. Il exploite plutôt les motifs présents dans les quantités de lectures intra-répétitives (IRR) (Figure 1) pour identifier les valeurs aberrantes ou les différences de distribution entre cas et témoins. Ainsi, EHDn ne fonctionne qu’avec des régions plus longues que la longueur de lecture, car les allèles de répétitions pathogènes sont souvent des expansions et généralement longs (Dolzhenko et al., 2020). Cette approche rend inutile la constitution d’un catalogue de régions, ce qui facilite des découvertes impossibles à réaliser avec d’autres outils.
La performance de ces approches influe directement sur leur utilité et leur applicabilité. gangSTR est beaucoup plus rapide qu’EH (Mousavi et al., 2019) et, par conséquent, son catalogue comprend environ 830 000 loci, contre seulement 30 pour EH. Cela permet d’utiliser gangSTR pour le profilage du génome entier, une opération informatiquement irréalisable avec EH, et facilite la découverte de nouvelles associations et les études d’exploration non ciblées des STR.
Des outils de balayage génomique ont également été développés pour les données de séquençage à longues lectures. RepeatHMM-scan (Liu, Tong et Wang, 2020) peut utiliser les données ONT ou PacBio pour analyser le génome entier d’un échantillon et estimer le nombre de répétitions. Lors de mes tests, j’ai cependant constaté qu’il reste trop lent pour effectuer un balayage aussi précis au niveau de l’échantillon dans un délai comparable à celui de gangSTR.
Nous sommes aujourd’hui à l’ère du calcul de profils STR précis pour le génome entier, même pour les STR courts, le nombre de répétitions devenant moins précis à mesure que la région s’allonge. Étant donné que les STR longs ont tendance à être associés à des phénotypes pathogènes, des recherches supplémentaires seront nécessaires pour élucider le lien complet entre les répétitions en tandem et leurs effets phénotypiques potentiels chez l’humain.
Quelles sont les prochaines étapes ?
Malgré la diversité des approches disponibles pour l’analyse STR, aucune solution actuelle ne correspond à l’idéal hypothétique d’un outil d’appel de séquences répétées en tandem (STR) capable de déterminer rapidement et précisément les génotypes de n’importe quelle région STR, de courte à très longue.
Actuellement, les outils existants ne permettent pas de déterminer précisément le nombre de répétitions STR longues, ou ne sont pas suffisamment rapides pour le faire sur un jeu de données de séquençage de génome entier. Un outil idéal combinerait une grande exactitude et une grande précision dans le comptage des répétitions avec une vitesse d’exécution extrêmement rapide, pour une utilisation dans les études de découverte et d’association sur tous les loci STR du génome.
Un outil d’appel de STR parfait ne nécessiterait pas de catalogue préexistant, permettant ainsi l’identification de nouvelles expansions et facilitant les migrations entre génomes de référence. Les catalogues peuvent être incomplets, notamment parce que le génome de référence standard actuel (GRCh38) comporte de vastes régions non résolues qui commencent seulement à être étudiées (voir Nurk et al., prépublication de 2021).
Tout outil performant d’analyse de données biologiques doit être rigoureusement validé à l’aide de jeux de données réels. Malheureusement, concernant les séquences répétées en tandem (STR), il existe actuellement un manque de jeux de données de référence (« vérité terrain ») pour la validation (Mousavi et al., 2019). Il est donc essentiel d’exploiter pleinement les ressources disponibles (outils existants, lectures simulées, panels forensiques, électrophorèse capillaire, etc.) et de développer de meilleures approches de validation.
Mousavi et al. suggèrent également qu’une approche hybride, combinant données de séquençage à courtes et longues lectures, pourrait permettre une évaluation plus précise des STR, en tirant parti de la structure supplémentaire…
Les longues séquences offrent un contexte plus riche et les courtes séquences présentent généralement un taux d’erreur plus faible. Cela pourrait se traduire par un outil entièrement nouveau ou par une approche consensuelle tirant parti des atouts variés des méthodes existantes.
Dans le cadre de mon projet avec le Dr Bourque au Centre de génomique de McGill, j’explore les outils STR existants et leur application à la caractérisation d’une cohorte québécoise. Mon objectif est de développer de nouvelles techniques pour mieux comprendre cette forme de variation génétique.
Références
- 1. Gymrek, M. et al. lobSTR : Un profileur de séquences répétées en tandem courtes pour les génomes personnels. Genome Res., 2012 https://dx.doi.org/10.1101/gr.135780.111
- 2. Gymrek, M. et al. Contribution importante des séquences répétées en tandem courtes à la variation de l’expression génique chez l’humain. Nat. Genet., 2016. https://dx.doi.org/10.1038/ng.3461
- 3. Maladie de Huntington. MedlinePlus, 2020. https://medlineplus.gov/genetics/condition/huntington-disease/
- 4. Benson, G. Tandem repeats finder : un programme d’analyse des séquences d’ADN. Nucleic Acid Res., 1999. https://dx.doi.org/10.1093/nar/27.2.573TRF
- 5. Dolzhenko, E. et al. ExpansionHunter Denovo : une méthode informatique pour localiser les expansions de répétitions connues et nouvelles dans les données de séquençage à courte lecture. Genome Biology, 2020. https://dx.doi.org/10.1186/s13059-020-02017-z
- 6. Dolzhenko, E. et al. Détection d’expansions de longues répétitions à partir de données de séquençage de génomes entiers sans PCR. Genome Res., 2017 https://dx.doi.org/10.1101/gr.225672.117
- 7. Mousavi, N. et al. Profilage du paysage génomique des expansions de répétitions en tandem. Nucleic Acid Res., 2019 https://doi.org/10.1093/nar/gkz501
- 8. Illumina. Longueur maximale de lecture pour les plateformes de séquençage Illumina. 2020. https://emea.support.illumina.com/bulletins/2020/04/maximum-read-length-for-illumina-sequencing-platforms.html
- 9. Liu, Q., Tong, Y., et Wang, K. Détection à l’échelle du génome des expansions de séquences répétées en tandem courtes par séquençage à longues lectures. BMC Bioinformatics, 2020. https://dx.doi.org/10.1186/s12859-020-03876-w
- 10. Nurk, S. et al. La séquence complète d’un génome humain. Prépublication BioRxiv, 2021.https://dx.doi.org/10.1101/2021.05.26.445798v1
- 11. Amarasinghe, S. et al. Opportunités et défis de l’analyse des données de séquençage à longues lectures. Genome Biology, 2020.https://dx.doi.org/10.1186/s13059-020-1935-5
Publié le 28 juillet 2021