
Whole Genome STR Analysis
About Short Tandem Repeats
In early 2021, Jeffrey Hyacinthe, another student here in Guillaume Bourque’s lab, wrote about repetitive sequences, focusing on transposable elements (TEs). Here I will discuss another type of repetitive sequence: short tandem repeats (STRs), also known as microsatellites. These do not move around like TEs, and they contain multiple copies of short motifs 2-6 base pairs long (Gymrek et al., 2012; 2016). The number of repeats at a location can vary within an individual or population. There are estimated to be ~700 000 STRs in the human genome (Gymrek et al., 2016) meaning they are quite common. They constitute the main topic for my M.Sc. thesis research, commenced last Fall in the Bourque lab.
STRs have been associated with a variety of disease phenotypes such as Huntington’s Disease (HD) and Fragile X Syndrome (FXS), as well as other effects like expression variation (Gymrek et al., 2016). In HD, an expanded, in-frame C-A-G repeat in the protein-coding HTT gene triggers the onset of the disorder prior to the individual’s 30’s or 40’s (MedlinePlus, 2020; see Table 1). HD is associated with motor dysfunction and cognitive impairment, eventually resulting in death. Only one copy of the pathogenic allele is needed to cause disease onset.
| Number of Repeats | Phenotype |
|---|---|
| 10-35 | No phenotype |
| 36-39 | Potential onset of HD |
| 40+ | Almost guaranteed onset of HD |
Table 1: Repeat counts of CAG in the HTT gene (GRCh38 coordinates: chr4:3074876-3074933) and associated phenotypes. See MedlinePlus, 2020.
Researchers have put significant amounts of work into annotating repetitive regions in current reference genomes. Tandem Repeats Finder, originally published in 1999 by Gary Benson, is used to catalog ‘simple’ repeats in reference genomes – these annotations can be used by other tools as targets for genotyping or expansion detection (Mousavi et al., 2019).
Even with annotations, repetitive DNA is difficult to analyze with the genomic technologies currently available to us. One of the de-facto standard sequencing technologies in 2021 is the Illumina short-read sequencing platform (Amarasinghe et al., 2020), which typically produces paired reads of up to ~150 base pairs long (Illumina, 2020). To identify repetitive regions longer than this, a fairly obvious problem emerges (Figure 1).
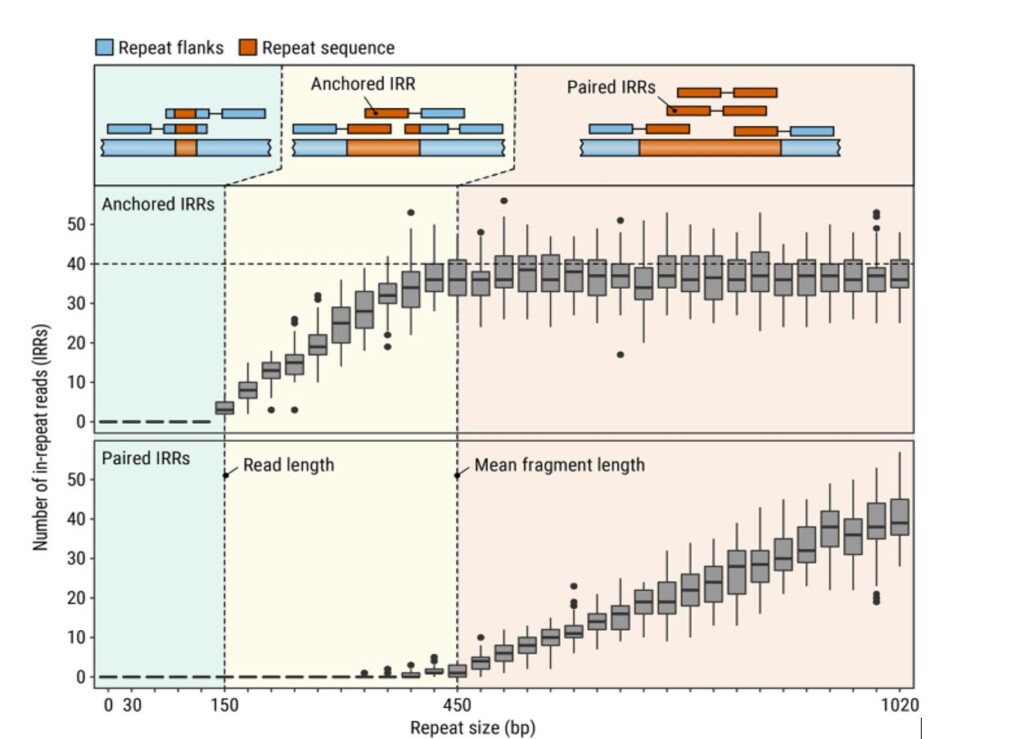
Figure 1: Types of read pairs from Illumina sequencing and how they align to different repeat sizes. Taken from Dolzhenko et al., 2020. The diagram shows that for repetitive sequences longer than 150bp, we no longer have reads that flank both sequences’ ends, and we start having to rely on approximate methods using in-repeat reads (IRRs) to determine repeat count. Paired IRRs can potentially map to multiple regions in the reference genome, since they have no anchoring sequence.
Long stretches of the same motif do not necessarily uniquely occur at only one position in the human genome (Dolzhenko et al., 2017). Without broader context of the surrounding regions, reads from one locus can potentially map to other loci, reducing their usefulness.
Adding more complexity to the calling process, we are primarily diploid creatures – most repeat loci have two copies in the human genome. If these loci are large or if we do not have enough sequencing coverage in an area, calling can become ambiguous and we may experience allelic dropout or have other difficulties determining a precise genotype (Gymrek et al., 2012; 2016; Mousavi et al., 2019).
Other techniques that show promise for STR genotyping are based on long-read sequencing technologies such as Oxford Nanopore (ONT) or Pacific Biosciences (PacBio) SMRT sequencing. Long reads are typically on the order of kilobases long (Amarasinghe et al., 2020), giving information on overall structure each read and eliminating the read-mapping issues presented above, except in extreme cases. This comes at the cost of higher sequencing prices, generally lower coverage, and higher error rates (Amarasinghe et al., 2020). Comparatively few tools have been developed for targeted genotyping and genome-wide scanning of STRs using long-read data.
Technologies to ascertain STRs
Specific tools are used to profile STR variation and call genotypes. When calling an STR genotype, we typically use the number of repeats of the motif unit to represent a particular allele. For the HTT repeat, a genotype of 20/35 would represent 20 and 35 copies of CAG, or 60 and 105 bases, on the two copies of chromosome 4 in the individual.
Some of the earliest of these computational methods which operate on NGS reads were released in the early 2010s. One of the first examples is lobSTR (Gymrek et al., 2012), which was designed to work on high-throughput sequencing data of the era and was validated against traditional DNA electrophoresis techniques. Some other STR-focused tools introduced over the past decade include hipSTR, Tredparse, and STRetch. These rely on catalogs and generally cannot detect the repeat counts of any STR longer than an Illumina read or only report expansions (Mousavi et al., 2019).
More recently, tools like ExpansionHunter (EH; Dolzhenko et al., 2017) and gangSTR (Mousavi et al., 2019) have developed new techniques which yield a greater number of higher quality calls, faster performance, and the ability to call confidence intervals on repeat regions that are longer than the read lengths themselves. These techniques use a mix of anchored/flanking reads and in-read repeats (see Figure 1) to both precisely call shorter STRs and call longer STR regions in a ‘fuzzy’ manner.
An alternative approach to using short reads to analyze long STR regions was introduced by the authors of EH in 2020 under the name ExpansionHunter Denovo (EHDn; Dolzhenko et al., 2020). This tool is not a caller at all, as it does not attempt to discern the number of repeats at a given locus. Instead, it takes advantage of patterns within in-repeat read (IRR) quantities (Figure 1) to look for outliers or case/control distribution differences. This means that EHDn only functions with regions longer than the read length – relying on pathogenic repeat alleles often being expansions, and generally long (Dolzhenko et al., 2020). This approach means a catalog of regions is unnecessary, which facilitates discoveries that would be impossible to make with other tools
The performance of these approaches has a direct impact on their usefulness and applicability. gangSTR is much faster than EH (Mousavi et al., 2019), and as a result includes a catalog of ~830 000 loci versus EH’s catalog of only 30 loci. This allows gangSTR to be used for whole-genome profiling, something which is computationally infeasible with EH, and facilitates new association discoveries and non-targeted STR exploration studies.
Genome-wide scanning tools have also been developed for long-read sequencing data. RepeatHMM-scan (Liu, Tong, & Wang; 2020) can use ONT or PacBio data to scan a sample’s whole genome to estimate repeat counts. Testing it myself, however, I found that it is still too slow to scan at sample level detail within the same order-of-magnitude time frame as gangSTR.
We are at a time where precise whole-genome STR profiles are possible to compute for short STRs, with repeat counts becoming fuzzier the longer a region gets. Given that longer STRs tend to be associated with pathogenic phenotypes, further work will be required to elucidate the complete link between tandem repeats and putative phenotypic effects in humans.
Where do we go from here?
Despite a variety of approaches being available for STR analysis, no one solution currently approaches a hypothetical “ideal” tandem repeat caller which quickly and accurately resolves the genotypes of any (short to very long) tandem repeat region.
Currently, existing tools are unable to precisely determine long STR repeat counts or are not fast enough to do this across a whole genome sequencing dataset. An ideal tool would combine high accuracy and precision of these repeat counts with extremely fast performance, for use in discovery and association studies across all STR loci in the genome.
A perfect STR caller would not require a pre-existing catalog, permitting the identification of novel expansions and allowing for easy migrations between reference genomes. Catalogs can be incomplete, especially since the current standard reference genome (GRCh38) has large unresolved regions which are only recently starting to be addressed (see Nurk et al., 2021 preprint).
Any high-quality tool analyzing biological data needs to be extensively validated with real world datasets. An unfortunate reality of tandem repeats is that there is currently a lack of “ground truth” datasets to validate against (Mousavi et al., 2019) – so it is essential to fully use what is available to us (existing tools, simulated reads, forensic panels, capillary electrophoresis, etc.) and developing better validation approaches.
Mousavi et al. also suggest that a hybrid approach utilizing both short- and long-read data may yield the most accurate assessment of STRs, taking advantage of the additional structural context long reads provide and short reads’ typically lower error rate. This could take the form of a completely new tool, or it could be a consensus-based approach which uses the varied strengths of existing methods.
As part of my project with Dr. Bourque here at the McGill Genome Centre, I am exploring existing STR tools and how they can be used to characterize a Québec cohort. I hope to develop new techniques to better understand this form of genetic variation.
References
- 1. Gymrek, M. et al. lobSTR: A short tandem repeat profiler for personal genomes. Genome Res., 2012 https://dx.doi.org/10.1101/gr.135780.111
- 2. Gymrek, M. et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet., 2016. https://dx.doi.org/10.1038/ng.3461
- 3. Huntington disease. MedlinePlus, 2020. https://medlineplus.gov/genetics/condition/huntington-disease/
- 4. Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acid Res., 1999.https://dx.doi.org/10.1093/nar/27.2.573TRF
- 5. Dolzhenko, E. et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biology, 2020. https://dx.doi.org/10.1186/s13059-020-02017-z
- 6. Dolzhenko, E. et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res., 2017 https://dx.doi.org/10.1101/gr.225672.117
- 7. Mousavi, N. et al. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acid Res., 2019 https://doi.org/10.1093/nar/gkz501
- 8. Illumina. Maximum read length for Illumina sequencing platforms. 2020. https://emea.support.illumina.com/bulletins/2020/04/maximum-read-length-for-illumina-sequencing-platforms.html
- 9. Liu, Q., Tong, Y., and Wang, K. Genome-wide detection of short tandem repeat expansions by long-read sequencing. BMC Bioinformatics, 2020. https://dx.doi.org/10.1186/s12859-020-03876-w
- 10. Nurk, S. et al. The complete sequence of a human genome. BioRxiv preprint, 2021.
https://dx.doi.org/10.1101/2021.05.26.445798v1 - 11. Amarasinghe, S. et al. Opportunities and challenges in long-read sequencing data analysis. Genome Biology, 2020.
https://dx.doi.org/10.1186/s13059-020-1935-5
Published: July 28 2021