
How do jumping genes contribute to human diversity?
In recognition of Transposons Day 2021 (June 16th), let’s take a look at the importance of active mobile genomic elements (once called “junk DNA”) to human health and diseases.
Illustration: ANDRZEJ KRAUZE
We are hosts to genomic hitchhikers
Of the three billion base pairs of the human genome, more than half contain a heterogenous crowd of repeated sequences called transposable elements (TEs, or transposons). These genomic hitchhikers have fascinated researchers since their discovery in corn by Barbara McClintock in the late 1940’s (McClintock was eventually awarded the Nobel Prize for her discovery… in 1983). As a matter of fact, TEs can indeed “jump” – or transpose – from one locus to another, sometimes affecting the expression of nearby genes.
TE jumps are not benign, since new insertions can disrupt genes and other crucial regulatory features. Indeed, TE can cause diseases: In germ line cells, TE insertions can cause sporadic conditions, while in soma cells, TE jumps can lead to cancer and are associated with aging and neurodegeneration.
Fortunately, most of the TEs present in our genomes today are no less than DNA fossils, inert witnesses of past waves of transposition. In fact, most TEs in the human genome have either accumulated inactivated mutations or remain silent under the control of epigenetic defense mechanisms.
Because TEs carry their own genes (transposases, reverse transcriptases, envelopes) and regulatory sequences (promoters, binding motifs), many have also been domesticated (or “co-opted”) throughout the evolution of eukaryotes. In human lineage, this “cherry picking” of useful TEs has led to important innovations, for example in the immune system.
Given all these examples, the fate of a new TE insertion is often difficult to predict. In humans, the overwhelming majority of TEs are fixed in the global population, meaning that most TE loci are shared between humans – most, but not all!
So, what are the consequences of contemporary TE activity in humans? How many are there? What do they do? Why does it matter? Hang on!
Active Transposable Elements generate structural variation
In humans, a small number of elements which belong to the Alu, LINE1 and SVA superfamilies (groups of homologous copies) escape the host defenses and retain their “jumping” capabilities. These TEs belong to the class ofretrotransposons (Class I) since they all rely on the reverse transcription of their RNA to colonize new loci.
Active TEs in humans are estimated to generate ~1 new insertion every 50 births, contributing to more than 40,000 known insertions polymorphisms(even this is most likely a large underestimation). These structural variants recapitulates the genetic diversity oof the human population, as it can be observed with single nucleotide polymorphisms (SNPs).
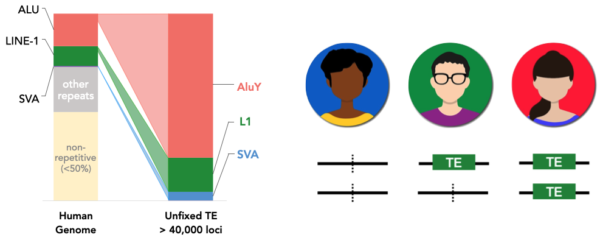
Figure 1. TE insertion polymorphism in humans accounts for tens of thousands of variants. If TE insertions present further analogies to SNPs, then a subset of TE variants should also be crucial in the regulation of gene expression. Understanding how TEs affect genomic regulation in humans is decisive, considering a growing number of evidences ties TE insertion polymorphisms to medical conditions.
Recent research, as discussed below, suggests that polymorphic TE insertions create functional variation among genomes and actively contribute to the emergence of new phenotypes.
New TE insertions can modulate gene expression
Expression QTL identifies hundreds of potential regulatory TEs
One popular approach to investigating the link between genomic variation and gene expression is to perform population-wide analyses called molecular QTL(quantitative trait loci). This new generation of QTL search for statistical connections between genotypes and molecular phenotypes throughout the genome. Molecular phenotypes are continuous variables typically drawn from -omics data, such as gene expression (eQTL), splice variants quantification (sQTL), chromatin accessibility (caQTL) or methylation (meQTL).
By swapping SNPs for TEs, researchers have recently been able to apply this framework to insertional polymorphisms of Alu, LINE1 and SVA among human genomes. The result? Hundreds of “TE-QTL”, where the presence of a given TE is statistically correlated to the expression of a nearby gene. First reported by Wang et al. (2017) in immune cell lines < a href=””>(LCL), the GTEx consortium recently showed that TE-QTL, like SNPs, can either promote tissue and organ-specific expression or display organism-wide (housekeeping) effects on gene expression. In addition, TE variants were shown to be able to generate splice variations after exploring TE-QTL in 44 post-mortem tissues.
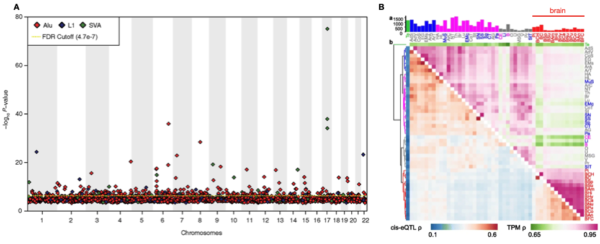
Figure 2. Recent findings using TE-eQTL. A Manhattan plot from Wang et al., (2017) displaying the significance of hundreds of Alu, L1 and SVA TE-eQTL, according to TE genomic location. B Heatmap of TE-eQTL (blue-red) and gene expression (green-pink) correlations across 44 tissues from the GTEx consortium. Adapted from Cao et al., (2020).
Leveraging multi-omics data to infer mechanisms
To understand how new TE insertions modulate nearby gene expression, I took advantage of the TE-QTL framework to layer epigenomic data and examine whether gene regulation by TEs could occur through chromatin remodeling. Using ATAC-seq, I tested this hypothesis by applying both expression and chromatin accessibility QTL (e- and ca-QTL) in LCLs derived from the 1000 Genomes Project.
This study found that hundreds of TEs are statistically associated with chromatin accessibility in humans, and that a subset of these elements also affect the expression of local genes. A great example of this relationship is the case of MAP3K13, an up-regulator of the proto-oncogene c-Myc for which both chromatin accessibility and gene expression are reduced in the presence of an AluYb8 insertion within an annotated enhancer. Though further investigation is needed, this example illustrates the potential of TEs to provide protective alleles by generating epigenomic variation.
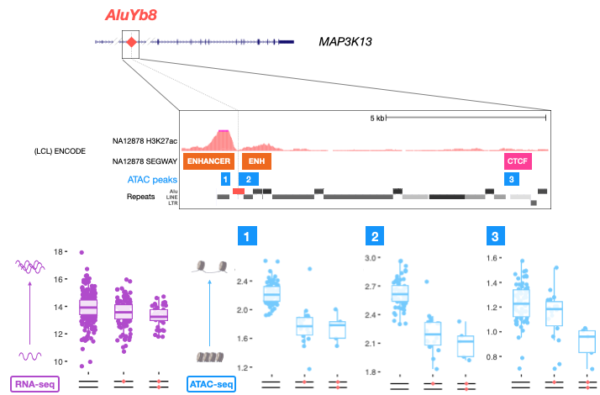
Figure 3. Figure 3. A candidate AluYb8 insertion in an enhancer of MAP3K13 is correlated to the reduction of chromatin accessibility at three ATAC-seq peaks mapped to TSS and CTCF sites (ATAC peaks 1, 2 and 3 and blue boxplots), as well as the reduction in expression of the gene as a whole (as seen by RNA-seq, purple boxplot). Adapted from Goubert et al., (2020a).
The importance of genotype quality
A critical aspect in all association studies (studies relying heavily on data correlations such as GWAS or QTL) is the quality of the genotypes. Bias in the initial genotyping can lead to missed or false positive signals during functional analyses.
While most genotyping algorithms rely on likelihood ratios after mapping reads onto a reference genome (an approach well-suited for SNPs) structural variants like TEs will only be represented by either presence or absence alleles. Given that the majority of datasets (large cohorts in particular) are still relying on reads shorter than a typical TE (active human TEs range from 300bp to 6000bp), there is a pressing need to improve TE genotyping.
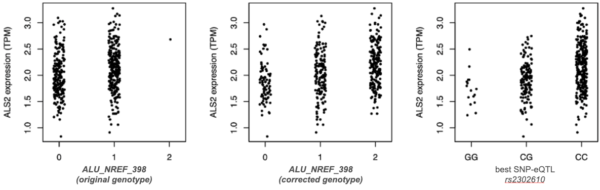
Figure 4. Errors in TE genotyping reduce the ability to detect TE-eQTL. In this example, only 1 homozygote for the TE insertion (“2”) was detected with a method based on a single reference genome (left, Sudmant et al,. 2015). Correction using a composite reference genome made of pairs of presence/absence alleles for each locus (centre) enhances the ability to detect a correlation between genotypes and ALS2 expression, as recapitulated by a SNP in linkage disequilibrium (right).
At the McGill Genome Centre, we develop new methods to improve TE genotyping and obtain a better understanding of their effects in humans:
- With short reads, TE genotypes can be improved by remapping reads over a composite genome made of pairs of “presence” and “absence” alleles using the linear reference genome as a background (project homepage, in development).
- If long reads and alternate genome assemblies are available, then personalized and graphed genomes are the next avenue to explore.
References:
- McClintock B., The origin and behavior of mutable loci in maize – Proceedings of the National Academy of Sciences Jun 1950, 36 (6) 344-355; DOI: 10.1073/pnas.36.6.344
- Payer, L.M., Burns, K.H. Transposable elements in human genetic disease – Nat Rev Genet 20, 760–772 (2019). DOI: 10.1038/s41576-019-0165-8
- Burns, K.H., 2017. Transposable elements in cancer. Nature Reviews Cancer, 17(7), pp.415-424.
- Andrenacci, D., Cavaliere, V. and Lattanzi, G., 2020. The role of transposable elements activity in aging and their possible involvement in laminopathic diseases. Ageing research reviews, 57, p.100995.
- Jönsson, M.E., Garza, R., Johansson, P.A. and Jakobsson, J., 2020. Transposable elements: a common feature of neurodevelopmental and neurodegenerative disorders. Trends in Genetics.
- Smit, A.F., Riggs A.D., Tiggers and DNA transposon fossils in the human genome – Proceedings of the National Academy of Sciences Feb 1996, 93 (4) 1443-1448; DOI: 10.1073/pnas.93.4.1443
- Slotkin, R., Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8, 272–285 (2007). DOI: 10.1038/nrg2072
- Cosby, R.L., Judd, J., Zhang, R., Zhong, A., Garry, N., Pritham, E.J. and Feschotte, C., 2021. Recurrent evolution of vertebrate transcription factors by transposase capture. Science, 371(6531).
- Chuong, E.B., Elde, N.C. and Feschotte, C., 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nature Reviews Genetics, 18(2), p.71.
- Feusier, J., Watkins, W.S., Thomas, J., Farrell, A., Witherspoon, D.J., Baird, L., Ha, H., Xing, J. and Jorde, L.B., 2019. Pedigree-based estimation of human mobile element retrotransposition rates. Genome research, 29(10), pp.1567-1577.
- Watkins, W.S., Feusier, J.E., Thomas, J., Goubert, C., Mallick, S. and Jorde, L.B., 2020. The Simons Genome Diversity Project: a global analysis of mobile element diversity. Genome biology and evolution, 12(6), pp.779-794.
- Cao, X., Zhang, Y., Payer, L.M., Lords, H., Steranka, J.P., Burns, K.H. and Xing, J., 2020. Polymorphic mobile element insertions contribute to gene expression and alternative splicing in human tissues. Genome biology, 21(1), pp.1-19.
- Goubert, C., Zevallos, N.A. and Feschotte, C., 2020a. Contribution of unfixed transposable element insertions to human regulatory variation. Philosophical Transactions of the Royal Society B, 375(1795), p.20190331
- Li, H., 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27(21), pp.2987-2993.
- Goubert, C., Thomas, J., Payer, L.M., Kidd, J.M., Feusier, J., Watkins, W.S., Burns, K.H., Jorde, L.B. and Feschotte, C., 2020b. TypeTE: a tool to genotype mobile element insertions from whole genome resequencing data. Nucleic acids research, 48(6), pp.e36-e36.
- Groza, C., Kwan, T., Soranzo, N., Pastinen, T. and Bourque, G., 2020. Personalized and graph genomes reveal missing signal in epigenomic data. Genome biology, 21, pp.1-22.
Published: June 21 2021