
Disambiguating mixed-species of graft samples
As a Bioinformatician, often I get to work with PDX cancer samples. I’ve recently been reading about samples containing genome admixture, and was revisiting strategies that we commonly use for analyzing these biological data. Presented here is a summary of the existing software tools used for this purpose.
What are Xenografts?
Studying and understanding cancer is very challenging, and animal model systems help in addressing some of the common research bottlenecks. Mouse harboring human cancer cells, also known as Patient-derived xenograft models (PDX), are an excellent model system available to researchers. A small number of cancer cells collected from a patient are injected into an immunocompromised mouse, which grow to form tumors. PDX systems provide a controlled platform to study tumor biology and are especially useful for testing chemotherapeutic approaches.
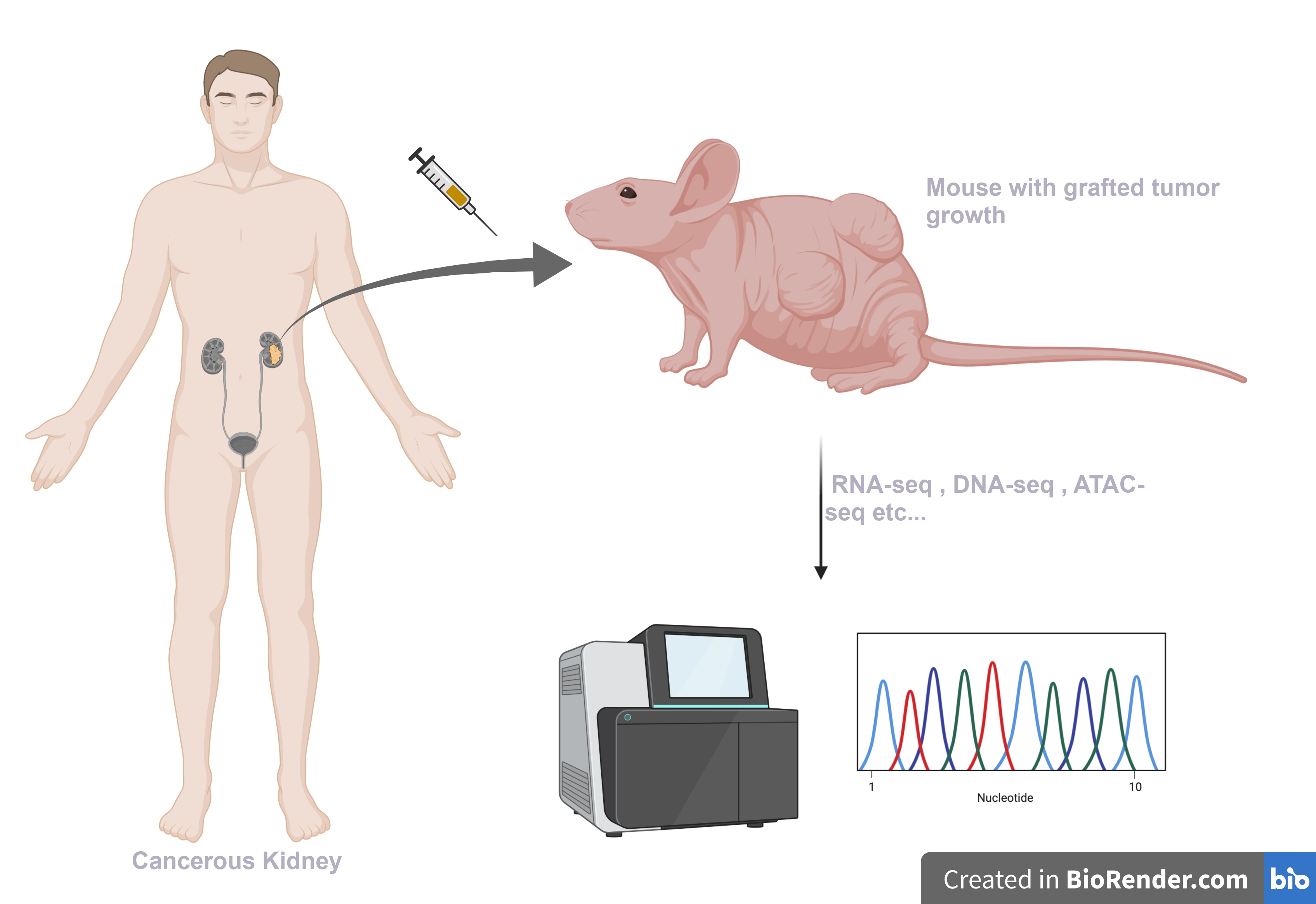
Technical issue with xenograft samples
The grafted sample obtained from these mouse tumors can be subjected to NGS for genomics and transcriptomics studies. Despite meticulous efforts, it is difficult to prevent contamination of the graft samples with the host (mouse) stromal tissue, and the sequencing obtained is usually contaminated with host DNAs and RNAs. These contaminations could hinder correct interpretation of the data. Removing reads of host origin before downstream analysis is becomes essential to ensuring accurate conclusions.
What are the methods available?
Various algorithms exist to separate host-derived reads from the rest of the sample. Almost all methods require that the input is in two are more BAM files: one aligned to the host genome and other aligned to the graft genome. The type of aligner used also hugely influences the choice of algorithm. I was able to find five well documented software packages. Most of these algorithm compare the quality of read alignment and then categorises the reads to either host or graft. If ambiguous the read is discarded. Some of the key features of these packages are highlighted in the table. (Table 1)
Table 1.
| Package/software Name | Compatible Aligner | Comparison | Remarks | Multicore | References |
|---|---|---|---|---|---|
| Sargasso | Bowtie2, STAR | Multispecies | Custom filtering by threshold | Yes | [1] |
| Xenosplit | Subread, Bowtie2, Subjunc, TopHat2, BWA and STAR | Maximum two species | Goodness of mapping scores | No | [2] |
| Disambiguate | Hisat2, TopHat, BWA and STAR | Maximum two species | No | [3] | |
| XenoCP | BWA | Maximum two species | Cloud-based | Yes | [4] |
I tried all three of the four of these packages (XenoCP omitted) in an active project. I had selected a sample dataset that had issues with poor read alignment to reference genome (GRch38). The reason I chose this dataset was to see if the unaligned reads are of host origin! But that was not the case for this dataset. I used the number reads recovered (assigned as graft-origin) as a parameter to compare the tools. Sargosso and xenosplit perform very similarly and are stringent in assigning the reads to graft. Disambiguate, the oldest program of those tried, gave slight improvement compared to standard reference genome-based alignment alone. In the future, I plan to compare these five packages using a synthetic dataset with known portion of mouse and human reads. Until then, Sargasso and xenosplit seem promising if you are interested in specificity and not sensitivity.
Table 2.
| Sample 1 | Sample 2 | Sample 3 | Sample 4 | |
|---|---|---|---|---|
| raw_reads | 187, 299, 238 | 140,927,692 | 328,021,530 | 342,910,322 |
| trimmed_reads | 187,126,248 | 140,809,146 | 327,809,782 | 342,667,064 |
| sargasso_Human (%) |
36,469,134 (19.49) |
261,686 (0.19) |
55,689,176 (16.99) |
18,074,244 (5.27) |
| xenosplit_Human (%) |
35,751,426 (19.11) |
361,651 (0.19) |
54,616,560 (16.99) |
18,028,870 (5.27) |
| disambiguate_Human (%) |
46,050,656 (26.61) |
12,384,076 (8.79) |
82,209,686 (25.09) |
41,830,148 (12.21) |
| GRCh38(unique) (%) |
51,406,614 (24.47) |
13,458,944 (9.56) |
83,642,740 (25.52) |
49,287,080 (14.38) |
Reference:
Qiu, J., et al., Mixed-species RNA-seq for elucidation of non-cell-autonomous control of gene transcription. Nature Protocols, 2018. 13(10): p. 2176-2199.
Giner, G. and A. Lun, https://github.com/goknurginer/XenoSplit. 2019.
Ahdesmaki, M.J., et al., Disambiguate: An open-source application for disambiguating two species in next generation sequencing data from grafted samples. F1000Res, 2016. 5: p. 2741.
Rusch, M., et al., XenoCP: Cloud-based BAM cleansing tool for RNA and DNA from Xenograft. bioRxiv, 2020: p. 843250.